
PLEASE NOTE:
We are currently in the process of updating this chapter and we appreciate your patience whilst this is being completed.
Patterns of inheritance
Whether a child ends up with a medical condition depends on several factors, such as what genes they inherit, whether the gene they inherit for a condition is dominant or recessive and their environment. Pedigree charts allow us to follow the history in a family of a genetic condition and draw conclusions about inherited traits as illustrated in the figure below.
Figure 2: Determining Inheritance from Pedigree Charts
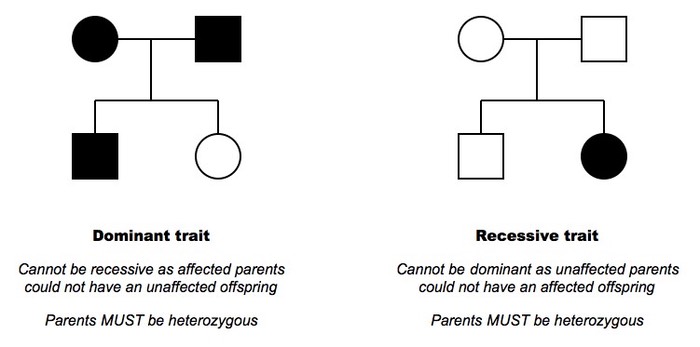
http://ib.bioninja.com.au/standard-level/topic-3-genetics/34-inheritance/pedigree-charts.html
Monogenetic disorders
Monogenetic disorders result from a mutation in a single gene and are relatively rare. Typically, children inherit the gene – very rarely the mutation arises spontaneously. Monogenetic disorders are defined by the chromosome type on which the mutation occurs and can either be dominant or recessive:
In autosomal recessive conditions, the individual develops the disease only if they inherit two copies of the mutant allele. They must therefore inherit a copy from each parent. If the child only inherits one copy, then they will be a carrier, but will be phenotypically normal. A carrier has a 50% chance of passing on the mutant allele to each child. If both parents are carriers, then there is a 25% chance of the child inheriting both copies and being born with the disease. Examples of autosomal recessive conditions include cystic fibrosis (condition in which lungs become clogged with thick mucus), sickle cell anaemia (abnormal development of red blood cells) and Tay-Sachs disease (a progressive nervous system disease).
In autosomal dominant conditions, an individual can develop the disease with either one or two copies of the mutant allele. There is, therefore, no carrier state as all those with the gene have an abnormal phenotype. A person with one parent who has the disease has a 50% chance of inheriting the mutant gene. Examples of autosomal dominant conditions include Huntington’s disease (a cause of premature dementia) and a form of polycystic kidney disease (a condition resulting in cyst development in kidneys).
Sex-linked disorders occur when the gene associated with the disease is on one of the sex chromosomes, almost invariably the X-chromosome. They are usually inherited recessively. Women are more likely to be carriers of X-linked recessive conditions by nature of having two X-chromosomes and men are more likely to develop disease. All daughters of affected men are therefore carriers and no sons are affected due to inheriting the Y chromosome. When a women is a carrier, her daughters have a 50% chance of becoming a carrier. Examples of sex-linked disorders include Duchenne muscular dystrophy (a cause of progressive muscle weakness) and haemophilia (a disorder of blood coagulation). X-linked dominant and Y-linked conditions are extremely rare. Examples include Rett syndrome and male infertility respectively.
Mitochondrial genetic conditions are maternally inherited, with mutations typically resulting in energy deficiency in cells as seen with Leigh’s disease. There is no predictable inheritance pattern and the severity of mitochondrial diseases can vary according to the proportion of mitochondria that carry the mutation. This is known as the threshold effect – a certain proportion of mutated mitochondrial DNA is required to be inherited for the individual to develop symptoms of the disease.
Polygenic disorders
Polygenic disorders are caused by several gene variants, each affecting susceptibility to disease. There is increasing evidence to suggest strong genetic components to disorders not previously thought to be genetic and they are, therefore, likely to be common in the population, much more so than single gene disorders. Examples of diseases influenced by many genes include cancer, coronary heart disease and diabetes. For diabetes, if an individual’s mother or father has type 1 diabetes, the individual’s risk is increased by 2% and 8% respectively. If either the mother or the father has type 2 diabetes, an individual’s risk is increased by 15%. Identifying these genes can help improve understanding of disease aetiology, help predict who is at risk, assist with targeting disease prevention and treatment, and help identify new targets for therapies. However, due to complex inheritance patterns, it can be difficult to identify these genes.
Chromosomal disorders
Chromosomal disorders can result from numerical or structural abnormalities and can be inherited or appear de novo, with the latter generally more common. Microscopic techniques are available to assess the number and structure of chromosomes (e.g. Down syndrome), with a karyotype used to provide an organised picture of a person's chromosomes. The abnormalities usually result from an error in cell division, either in mitosis (where identical copies of the original cell are produced) or in meiosis (where gametes are produced), If they occur in the gametes then the abnormality will be present in every cell in the offspring, while if they happen after conception then only some cells will have the abnormality. Factors can increase the risk of this happening, such as increasing maternal age and environmental factors.
Penetrance
The penetrance of a genetic condition is the likelihood that a person carrying a disease-associated genotype will develop the disease. Single gene disorders have high penetrance; both Huntington's disease and cystic fibrosis are near 100% penetrant by the age of 70 and at birth respectively, meaning all affected individuals will develop the disease.
For those mutations which increase the risk of developing a disease, the risk is time-dependent, increasing over time. Mutations in the BRCA1 gene that are associated with familial breast cancer have a lifetime prevalence of 60-85%. That translates to 6-8.5 women out of 10 who have the mutation developing breast cancer during their lifetime. Women with BRCA mutations also have an increased risk of developing breast cancer at a younger age. A recent study found for BRCA1 carriers, the incidence of breast cancer per decade of age increased from 21-30yrs to 31-40yrs but then remained fairly consistent from 31-70yrs, with peak incidence in 41-50yrs. A similar pattern was seen for BRCA2 carriers, with peak incidence in 51-60yrs.
Conversely polygenic disorders tend to have low penetrance, with the likelihood of developing the disease affected by the modifying effects of other genes and/or environmental factors.
Gene-environment interactions
Most genetic disorders are multifactorial; they result from a complex interplay between inherited mutations in multiple genes, often interacting with environmental factors. The susceptibility to developing disease such as heart disease and diabetes can increase following exposures such as infections, chemicals, physical hazards, nutritional exposures and behaviours. An example is phenylketonuria: in the absence of dietary phenylalanine, no disease develops.
Study types/genetic epidemiology
Studying the inheritance patterns of genetic disorders can help target messages and interventions to people most likely to benefit. Genetic epidemiologists use various types of study design to investigate gene-disease relationships as summarised below:
Family studies can be used to determine initially if there is a genetic component to a disorder. If there is a higher risk of disease in family members of an affected person than in the general population, this suggests a genetic component.
The position on the chromosome implicated can be determined through linkage studies. Genes that are close together on the same chromosome are more likely than expected by chance to be inherited together as they are less frequently separated by recombination. The frequency with which two genes are inherited together or linked depends on the length of the DNA between the two genes. This linkage between genes then helps to locate the relative positions of genes on the chromosomes.
Association studies identify specific genes by measuring the relative frequency with which a specific polymorphism occurs with a disease of interest in the population. For example, genes encoding the cell-surface receptor molecule CCR5 have been shown to be associated with increased resistance to infection by HIV, and polymorphisms encoding the major histocompatibility complex play a major role in immune response to pathogens.
Genome-wide association studies look at genetic variants across the whole genome in different individuals to see if any variant is associated with a particular trait. These types of studies can find genetic variations that contribute to common complex diseases such as mental illness and asthma. Two groups of people provide DNA, one with the disease and one without the disease, provide DNA and markers of genetic variation, single nucleotide polymorphisms, are sought. If these are more common in the group with the disease, the variations are said to be associated with the disease, although they may not directly cause the disease. Further sequencing work is needed to identify the exact genetic change.
Other genetic epidemiology studies explore the relative contributions of genetic and environmental factors to the risk of developing disease. However, these types of studies are difficult and expensive to carry out for the following reasons:
- Large numbers of cases and controls are needed to obtain statistically robust results as there are often many genes increasing susceptibility, each of which make a small contribution. Therefore, a very large sample size is required to detect the effects of the genes.
- There will be heterogeneity within and between populations with different gene pools and varying environmental exposures.
- An incomplete understanding of the pathogenesis of the disease may mean that not all relevant genes in an individual are detected and may be missed. Certain genes are often selected as they code for biological functions connected with the disease. A genome-wide association study is then required.
Because of these difficulties, many associations have been reported but not independently verified.
© Public Health Genetics Unit 2006, H Green 2017