PLEASE NOTE:
We are currently in the process of updating this chapter and we appreciate your patience whilst this is being completed.
The prodominant study designs can be categorised into observational and interventional studies. Observational studies, such as cross-sectional, case control and cohort studies, do not actively allocate participants to receive a particular exposure, whilt interventional studies do. Each of the above study designs are described here in turn.
Cross-sectional Studies
In a cross-sectional study, data are collected on the whole study population at a single point in time to examine the relationship between disease (or other health-related outcomes) and other variables of interest (exposures).
Cross-sectional studies therefore provide a snapshot of the frequency of a disease or other health-related characteristics in a population at a given point in time. This methodology can be used to assess the burden of disease or health needs of a population, for example, and is therefore particularly useful in informing the planning and allocation of health resources.
Types of cross-sectional study
- Descriptive
A cross-sectional study may be purely descriptive and used to assess the frequency and distribution of a particular disease in a defined population. For example, a random sample of schools across London may be used to assess the burden or prevalence of asthma among 12- to 14-year-olds.
- Analytical
Analytical cross-sectional studies may also be used to investigate the association between a putative risk factor and a health outcome. However, this type of study is limited in its ability to draw valid conclusions about any association or possible causality because the presence of risk factors and outcomes are measured simultaneously. It is therefore not possible to confidently infer whether the disease or the exposure came first, so causation should always be confirmed by more rigorous studies. The collection of information about risk factors is also retrospective, running the risk of recall bias.
In practice cross-sectional studies often include an element of both types of design.
Issues in the design of cross-sectional surveys
- Choosing a representative sample
A cross-sectional study should be representative of the whole of the population, if the findings are to have external validity. For example, a study of the prevalence of diabetes among women aged 40-60 years in Town A should comprise a random sample of all women aged 40-60 years in that town. If the study is to be representative, attempts should be made to include hard-to-reach groups, such as people in institutions or the homeless, to avoid selection bias.
- Sample size
The sample size should be sufficiently large to estimate the prevalence of the conditions of interest with adequate precision. Sample size calculations can be carried out using sample size tables or statistical packages such as Epi Info. The larger the study, the less likely the results will be due to chance alone, but this will also have implications for cost.
- Data collection
As data on exposures and outcomes are collected simultaneously, specific inclusion and exclusion criteria should be established at the design stage, to ensure that those with the outcome are correctly identified. The data collection methods will depend on the exposure, outcome and study setting, but include questionnaires and interviews, as well as medical examinations. Routine data sources may also be used.
Potential bias in cross-sectional studies
Non-response is a particular problem affecting cross-sectional studies and can result in bias of the measures of outcome. This is a particular problem when the characteristics of non-responders differ from responders. Recall bias can occur if the study asks participants about past exposures.
Analysis of cross-sectional studies
In a cross-sectional study all factors (exposure, outcome, and confounders) are measured simultaneously. The main outcome measure obtained from a cross-sectional study is prevalence:

For continuous variables such as blood pressure or weight, values will fall along a continuum within a given range. Prevalence may therefore only be calculated when the variable is divided into those values that fall below or above a particular pre-determined level. Alternatively, mean or median levels may be calculated.
In analytical cross-sectional studies the odds ratio can be used to assess the strength of an association between a risk factor and health outcome of interest, provided that the current exposure accurately reflects the past exposure.
Strengths and weaknesses of cross-sectional studies
Strengths
- Relatively quick, cheap and easy to conduct (no long periods of follow-up).
- Data on all variables is only collected once.
- Able to measure prevalence for all factors under investigation.
- Multiple outcomes and exposures can be studied.
- The prevalence of disease or other health-related characteristics are important in public health for assessing the burden of disease in a specified population and in planning and allocating health resources.
- Good for descriptive analyses and for generating hypotheses.
Weaknesses
- Difficult to determine whether the exposure or outcome came first (there may be reverse causality – see chapter 12 “Association and Causation”)
- Not suitable for studying rare diseases or diseases with a short duration.
- As cross-sectional studies measure prevalent rather than incident cases, the data will always reflect determinants of survival as well as aetiology.1
- Unable to measure incidence.
- Associations identified may be difficult to interpret.
- Susceptible to biases such as responder bias, recall bias, interviewer bias and social acceptability bias.
Case Control Studies
In a case-control study the study group is defined by the outcome (e.g. presence of a disease), not by exposure to a risk factor. The study starts with the identification of a group of cases (individuals with a particular health outcome) in a given population and a group of controls (individuals without the health outcome) from the same population.
The prevalence of exposure to a potential risk factor is then compared between cases and controls. If the prevalence of exposure is more common among cases than controls, the exposure may be a risk factor for the outcome under investigation.
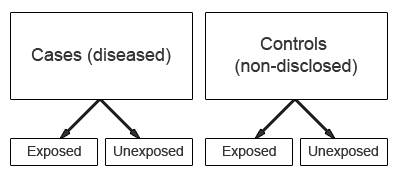
One of the advantages of case-control studies is that they can be used to study outcomes or diseases that are rare. However, a major characteristic is that data on potential risk factors are collected retrospectively and as a result may give rise to bias. This is a particular problem associated with case-control studies and therefore needs to be carefully considered during the design and conduct of the study.
Issues in the design of case-control studies
- Formulation of a clearly defined hypothesis
As with all epidemiological investigations the beginning of a case-control study should begin with the formulation of a clearly defined hypothesis.
- Case definition
It is essential that the case definition is clearly defined at the outset of the investigation to ensure that all cases included in the study are based on the same diagnostic criteria.
- Source of cases
The source of cases needs to be clearly defined. It is important to ensure that cases selected for a study are representative of all cases of the disease in the population. Cases may be recruited from a number of sources; for example, they may be recruited from a hospital, clinic, GP registers or population registries. Population-based case-control studies are generally more expensive and difficult to conduct, but are also more representative of the spectrum of disease.
- Selection of cases
Case-control studies may use incident or prevalent cases.Incident cases comprise cases newly diagnosed during a defined time period. The use of incident cases is considered to be preferable because the recall of past exposure(s) may be more accurate amongst those who have been recently diagnosed with a condition. In addition, the temporal sequence of exposure and disease is easier to assess among incident cases.
Prevalent cases comprise individuals who have had the outcome under investigation for some time. Inclusion of prevalent cases may mean the results of the study are more generalisable to the wider population. However, it may also give rise to recall bias as prevalent cases may be less likely to accurately remember past exposures. As a result, the interpretation of results based on prevalent cases may prove more problematic as it may be more difficult to ensure that reported events (exposure) relate to a time before the development of disease rather than being a consequence of the disease process itself. For example, individuals may modify their exposure following the onset of disease. Another disadvantage of sampling prevalent cases is the risk of preferentially including the milder cases which have a better survival; these may not be representative of all disease cases, and may have different levels of the exposure(s) of interest.
- Source and selection of controls
A particular problem inherent in case-control studies is the selection of a comparable control group. Controls are used to estimate the background prevalence of exposure in the population which gave rise to the cases. Therefore, the ideal control group would comprise a random sample from the general population that gave rise to the cases. However, this is not always possible in practice.The goal is to select individuals in whom the distribution of exposure status would be the same as that of the cases in the absence of an exposure-disease association. That is, if there is no true association between exposure and disease, the cases and controls should have the same distribution of exposure. To put it another way, controls should meet all the criteria for cases, apart from having the disease itself, so if the cases are women aged 50-70 years with breast cancer, the controls should be selected from a similar group who do not have breast cancer.
The source of controls is dependent on the source of cases. In order to minimise bias, controls should be selected to be a representative sample of the population which produced the cases. For example, if cases are selected from a defined population such as a GP register then controls should comprise a sample from the same GP register.
In case-control studies where cases are hospital based, it is common to recruit controls from the hospital population. However, the choice of controls from a hospital setting should not include individuals with an outcome related to the exposure being studied. For example in a case-control study of the association between smoking and lung cancer, the inclusion of controls being treated for a condition related to smoking (e.g. chronic bronchitis) may result in an underestimate of the strength of the association between the exposure (smoking) and outcome (lung cancer).
Recruiting more than one control per case may improve the statistical power of the study, especially where the number of cases is limited. However, there is little additional statistical power to be gained by recruiting more than 4 controls per case.
- Measuring exposure status
Exposure status is measured for each individual by assessing the level of exposure during the period of time prior to the onset of the condition under investigation, when the exposure could have acted as a causal factor.Note that in case-control studies the measurement of exposure is established after the development of disease and as a result is prone to both recall and observer bias.
Various methods can be used to ascertain exposure status. These include:
◦ Standardised questionnaires
◦ Biological samples
◦ Interviews with the subject
◦ Interviews with spouse or other family members
◦ Medical records
◦ Employment records
◦ Pharmacy records
The procedures used for the collection of exposure data should be the same for cases and controls.
Common sources of bias and confounding in case-control studies
Due to the retrospective nature of case-control studies they are particularly susceptible to the effects of bias which may be introduced as a result of a poor study design or during the collection of exposure and outcome data.
Because the disease and exposure have already occurred at the outset of a case-control study there may be differential reporting of exposure information between cases and controls based on their disease status. Cases and controls may recall past exposure differently, because knowledge of being a case may affect whether the individual remembers a certain exposure, for example (recall bias). Similarly, the recording of exposure information may vary depending on the investigator’s knowledge of an individual's disease status (interviewer/observer bias).
Selection bias can occur in case-control studies when the selected control group are not representative of the population from which the cases arose, thus comparisons of exposure distributions between cases and controls may give misleading results.
Temporal bias (also known as reverse causality) may also occur in case-control studies. When trying to establish a link between exposure and outcome, it must be clear that the exposure occurred well before the diagnosis of the disease of interest.
Therefore, the design and conduct of the study must be carefully considered as there are limited options for the control of bias during the analysis.
A confounder is a factor associated independently with both the exposure and outcome, and can be a problem where cases and controls differ with respect to a potential confounder. It can be dealt with at two stages:
- Design stage – when selecting controls, e.g. matching or restriction
- Analysis stage – statistical techniques including adjustment and multivariate techniques
More detail on adjusting for confounding can be found in the chapter “Bias and confounding”.
Analysis of case-control studies
The odds ratio (OR) is used in case-control studies to estimate the strength of the association between exposure and outcome. It is not possible to estimate the incidence or risk of disease from a case-control study, unless the study is population-based and all cases in a defined population are obtained.
The results of a case-control study can be presented in a 2x2 table as follows:
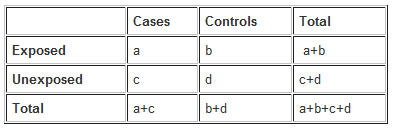
The odds ratio is a measure of the odds of exposure in the cases (= a/c), compared to the odds of exposure in the control group (= b/d). It is calculated as follows:
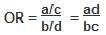
Example: Calculate the odds ratio from a hypothetical case-control study of smoking and pancreatic cancer among 100 cases and 400 controls, the results of which are shown below.
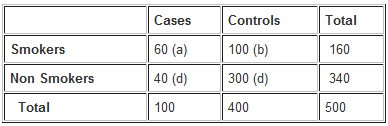
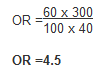
The OR calculated from the hypothetical data suggests that individuals with cancer of the pancreas (cases) are more likely to have smoked than those without the disease. Specifically, participants with pancreatic cancer have 4.5 times the odds of being smokers compared to those without pancreatic cancer.
NB: The odds ratio above has been calculated without adjusting for potential confounders. Further analysis of the data would involve stratifying by levels of potential confounders such as age. The 2x2 table can then be extended to allow stratum-specific rates of the confounding variables to be calculated and, where appropriate, an overall summary measure adjusted for the effects of confounding and a statistical test of significance. In addition, confidence intervals for the odds ratio would also be presented.
Strengths and weaknesses of case-control studies
Strengths
- Cost-effective relative to other analytical studies such as cohort studies.
- Case-control studies are retrospective, cases are identified at the beginning of the study therefore there is no long follow-up period (compared to cohort studies).
- Efficient for the study of diseases with long latency periods.
- Efficient for the study of rare diseases.
- Good for examining multiple exposures simultaneously.
Weaknesses
- Particularly prone to bias; especially selection, recall and observer bias.
- Case-control studies are limited to examining one outcome.
- Unable to estimate incidence rates of disease (unless study is population based).
- Poor choice for the study of rare exposures.
- The temporal sequence between exposure and disease may be difficult to determine.
Nested Case-control Studies
A nested case-control study is one where the cases and controls are selected from individuals within an established cohort study. The case-control study is thus said to be ‘nested’ within the cohort study.
Cases of a disease that arise within the defined cohort during the follow up period are identified, then a specified number of matched controls who have not developed the disease are selected from the same cohort. The main advantage of nested case-control studies is that certain exposure data will already have been collected for both cases and controls which limits the potential for recall bias.
Analysis is carried out in the same way as for normal case-control studies, with the calculation of odds ratios.
Strengths of nested case-control studies
- Relatively cheap and easy to conduct.
- Data related to exposure and confounding have often already been collected.
- Can utilise the baseline data on exposure and confounding collected before the onset of disease, which reduces the potential for recall bias and uncertainty regarding the temporal sequence between exposure and disease onset.
Weaknesses of nested case-control studies
- Causal inference is still limited.
- Not all relevant risk factor / exposure data may have been recorded.
- Data may not have been recorded in the same way, or with the same accuracy and consistency, over time.
- Recall bias may persist if some data are collected retrospectively.
- Non-diseased persons from whom the controls are selected may not be fully representative of the original cohort due to loss to follow-up or death.
Examples of case-control studies2
Case-control studies have been used in a variety of situations to evaluate possible causes of rare conditions. Classic examples include the investigation of cases of childhood leukaemia near the nuclear procession plant at Sellafield in Cumbria (UK), as well as cases of vaginal adenocarcinoma, which is normally rare, but were seen in higher numbers than usual in the USA in the 1970s. The difference between the young women with vaginal adenocarcinoma and their comparison group was that the mothers of cases had taken stilboestrol during the pregnancy (to prevent miscarriage), but the mothers of the controls had not.
- Gardner M J et al. Methods and basic data of case-control study of leukaemia and lymphoma among young people near Sellafield nuclear plant in West Cumbria. BMJ 1990; 300(6722):429-34
- Gardner M J et al. Results of case-control study of leukaemia and lymphoma among young people near Sellafield nuclear plant in West Cumbria. BMJ 1990; 300(6722): 423-9
- Herbst A, Ulfelder H, Poskanzer D. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. NEJM 1971; 284(15): 878-81
Cohort Studies
Cohort studies evaluate a possible association between exposure and outcome by following a group of exposed individuals over a period of time (often years) to see whether they develop the disease or outcome of interest. A cohort is a group of individuals who share a common characteristic, and may be chosen based on a population definition, or based on a particular exposure (see “Selection of study groups”, below). The incidence of disease in the exposed individuals of the cohort is then compared to the incidence of disease in unexposed, or lowest risk, individuals, and a relative risk (incidence risk or incidence rate) is calculated to assess whether the exposure and disease are associated.
Cohort studies may be prospective or retrospective, but both types define the cohort on the basis of exposure, not outcome.
Prospective cohort studies – participants are identified and followed up over time until the outcome of interest has occurred, or the time limit for the study has been reached. A temporal relationship between exposure and outcome can thus be established.
Retrospective cohort studies – exposure and outcome have already occurred at the start of the study. Pre-existing data, such as medical notes, can be used to assess any causal links, so lengthy follow-up is not required. This type of cohort study is therefore less time consuming and costly, but it is also more susceptible to the effects of bias. For example, the exposure may have occurred some years previously and adequate, reliable data on exposures may be differentially recorded in eventual cases compared to controls. In addition, information on confounding variables may be unavailable, inadequate or difficult to collect.
Issues in the design of cohort studies
- Selection of study groups
The aim of a cohort study is to select study participants who are identical with the exception of their exposure status. All study participants must be free of the outcome under investigation at the start of the study, and have the potential to develop the outcome under investigation.If the exposure is common, a defined study population can be selected for longitudinal assessment before classifying individuals as exposed or unexposed (population-based cohort study). This could involve, for example, a selection of the general population (e.g. defined by a geographical boundary) or all individuals who share a common workplace (occupational cohort).
If the exposure is rare, individuals may be chosen on the basis of exposure, to ensure sufficient exposed persons are enrolled. For example, this may be workers at a particular factory who regularly handle a chemical of interest. The comparison group might be workers at the same factory whose roles do not bring them into contact with the chemical. If a control group is chosen from the general population, there is a risk of bias due to the ‘healthy worker effect’: the general population is usually less healthy than the workforce because it includes those unable to work due to illness.3
- Measuring exposure
Levels of exposure (e.g. packs of cigarettes smoked per year) are measured for each individual at baseline at the beginning of study and assessed at intervals during the period of follow-up.A particular problem within cohort studies is determining whether individuals in the control group are truly unexposed. For example, study participants may start smoking after enrolment, or they may fail to correctly recall past exposure. Similarly, those in the exposed group may change their behaviour in relation to the exposure such as diet, smoking or alcohol consumption. The ability to repeatedly measure exposures over time, and to account for these, helps mitigate against such changes in behavior.
Exposure data may be obtained from a number of sources, including medical or employment records, standardised questionnaires, interviews and by physical examination.
- Measuring outcome
Outcome measures may be obtained from various sources including routine surveillance of cancer registry data, death certificates, medical records or directly from the participant. Note that the method used to ascertain outcome must be identical for both exposed and unexposed groups to avoid the risk of measurement bias. Information on outcomes should be collected by members of the study team who are blind to participants’ exposure status, to reduce the risk of observer bias.
- Methods of follow-up
The follow-up of participants in a cohort study is a major challenge, and it may take many years for a sufficient proportion of participants to have developed the outcome. A great deal of cost and time is required to ensure adequate follow-up of cohort members and to update measures of exposures and confounders, as well as monitoring participants’ health outcomes. Failure to collect outcome data for all members of the cohort will affect the validity of the study results.
Potential sources of bias in cohort studies
A major source of potential bias in cohort studies is losses to follow-up. Cohort members may die, refuse to continue participation in the study or fail to maintain contact. Such events may be related to the exposure, outcome or both, resulting in loss to follow-up bias. For example, individuals who develop a precursor to the outcome, such as symptoms of angina where the outcome of interest is a heart attack, may be less likely to continue to participate in the study. The degree to which losses to follow-up are correlated with either exposure or outcome can lead to significant bias in the measurement of the relationship between the exposure and outcome.1
Another source of potential bias in cohort studies arises from the degree of accuracy with which subjects have been classified with respect to their exposure or disease status. Differential misclassification – when one group of participants is more likely to have been misclassified than the other – can lead to an over- or underestimation of the relationship between the exposure and outcome.1
Analysis of cohort studies
The analysis of a cohort study uses the ratio of either the risk or rate of disease in the exposed cohort, compared with the risk or rate in the unexposed cohort.
If follow-up times differ markedly between participants, a rate may be more appropriate. The risk ratio uses as a denominator the entire group recruited at the start of the study while the rate ratio uses as a denominator the person-years which accounts for different lengths of time spent in the study. Different lengths of time spent in a study are due to varying start points, and incorporate multiple possible end points (i.e. one is no longer in the study on developing the outcome, or following death or moving away).
Calculation of the rate ratio from a hypothetical cohort study of smoking and pancreatic cancer followed-up for 1 year.
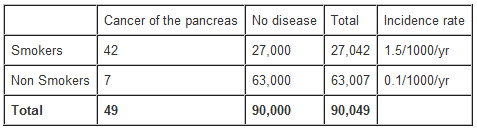
From the data in the table above, the rate ratio and attributable risk can be calculated as follows:

The rate ratio of 15 indicates that the risk of developing pancreatic cancer over 1 year is 15 times higher among smokers than non-smokers.
Attributable risk (AR)
AR = incidence rate among exposed (r1) – incidence rate among unexposed (r0)
AR = 1.5 – 0.1 = 1.4 /1000/yr
The attributable risk of pancreatic cancer due to smoking is 1.4 cases per 1000 per year
Attributable risk percentage (ARP), which gives the proportion of cases attributable (and thus avoidable) to an exposure in relation to all cases, is calculated as:
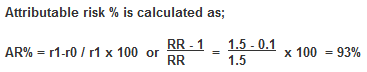
This can be interpreted as follows: smoking accounts for 93% of all cases of pancreatic cancer among smokers.
Standardised mortality and morbidity ratios (SMR) are another commonly used method of presenting results in a cohort study. See chapter “Numerators, denominators and populations at risk” for further information on SMR.
Strengths and weaknesses of cohort studies
Strengths
- Multiple outcomes can be measured for any one exposure.
- Can look at multiple exposures.
- Exposure is measured before the onset of disease (in prospective cohort studies) i.e. the temporal relationship is certain.
- Demonstrates direction of causality.
- Minimises selection bias (in prospective cohort studies)
- Good for measuring rare exposures, for example among different occupations.
- Good for outcomes which occur long after exposure (in retrospective cohort studies).
- Can measure incidence and prevalence.
Weaknesses
- Costly and time consuming.
- Prone to bias due to loss to follow-up.
- Knowledge of exposure status may bias classification of the outcome.
- Being in the study may alter participant behaviour.
- Inefficient for the study of a rare disease outcome.
- Classification of individuals (exposure or outcome status) can be affected by changes in diagnostic procedures.
- Cannot go back in time to collect information about exposure or confounders that were overlooked at the start of the study.
Examples of cohort studies2
One of the most famous examples of a cohort study is Sir Richard Doll’s study of the hazards of cigarette smoking in a cohort of nearly 35,000 British doctors. Baseline information about their smoking habits was obtained in 1951, and periodically thereafter. Cause-specific mortality was then monitored for 50 years and the results showed an excess mortality associated with smoking, chiefly due to vascular, neoplastic, and respiratory diseases.
Other cohorts include the Framingham cardiovascular studies, following people living in the town of Framingham, Massachusetts. Much of our current knowledge about heart disease, such as the effects of diet, exercise, and common medications such as aspirin, is based on this longitudinal cohort study. In the UK, the Whitehall studies have followed cohorts of British civil servants, demonstrating how groups in the cohort with differing levels of a characteristic, such as cholesterol, subsequently have different rates of ischaemic heart disease.
Cohort studies are also useful to study the longer-term effects of an unusual event. The most famous example of this is the cohort of people who survived the atomic bomb explosions at Hiroshima and Nagasaki.
- Doll R et al. Mortality in relation to smoking: 50 years' observations on male British doctors. BMJ 2004; 328 (7455): 1519
- E G Rael et al. Sickness absence in the Whitehall II study, London: the role of social support and material problems. Journal of Epidemiology and Community Health 1995:49 (5): 474-81
Intervention Studies (including Randomised Controlled Trials)
Intervention studies are designed to evaluate the effect of a specific treatment or practice and are considered to provide the most reliable evidence in epidemiological research.
Intervention studies can generally be considered as either preventative or therapeutic, and types of experimental intervention include:1
- Therapeutic agents
- Prophylactic agents
- Surgical procedures
- Health service strategies
Therapeutic trials are designed to evaluate the effect of therapies, such as new drugs or surgical procedures. They may also be known as clinical trials, and are conducted among individuals with a particular disease to assess the effectiveness of an agent or procedure in achieving a specific outcome, such as reduced mortality.1
Preventative trials are designed to evaluate whether an agent or procedure reduces the risk of developing a particular disease. They are carried out on individuals free from the disease at the beginning of the trial, but deemed to be at risk.1 Preventative trials may be conducted among individuals or entire communities, and examples include evaluations of new vaccines or bed nets to prevent infection with malaria.
Characteristics of an intervention study
- The intervention being tested is allocated to a group of two or more study subjects (individuals, households, communities).
- Subjects are followed prospectively to compare the intervention vs. the control (standard treatment, no treatment or placebo).
The main intervention study design is the randomised controlled trial (RCT).
Randomised controlled trials
The randomised controlled trial is considered the most rigorous method of determining whether a cause-effect relationship exists between an intervention and outcome.4 The strength of the RCT lies in the process of randomisation which is unique to this type of study design.
In an RCT, study participants are generally randomly assigned to one of two groups, the experimental group who will receive the intervention being tested, and a comparison group (controls) who receive a conventional treatment or placebo.4 These groups are then followed prospectively to assess the effectiveness of the intervention compared with the standard or placebo treatment.
Subjects are randomly allocated to the two study arms so that the intervention and control groups are as similar as possible in all respects, apart from the treatment. Potential confounding factors should be equally distributed between the two groups. The choice of comparison treatments may include an existing standard treatment, no treatment, an alternative treatment or a placebo. A placebo is a substance that resembles the intervention treatment in all respects except that it contains no active ingredients.
Figure 1. General outline of a two-arm randomised controlled trial.
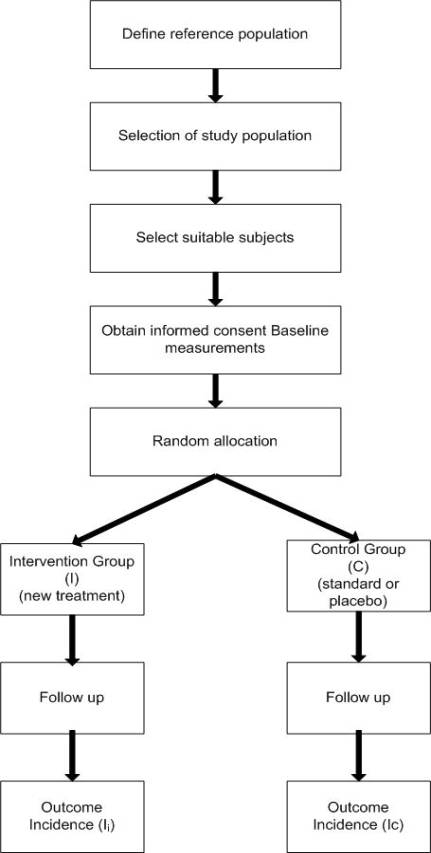
Basic outline of the design of a randomised controlled trial
Prior to the conduct of an RCT, a comprehensive study protocol should be developed and registered with a trials database, such as ClinicalTrials.gov. The study protocol will include information on:
- Aim and rationale of the trial
- Definition of the hypothesis
- Background/review of published literature
- Treatment schedules, dosage, toxicity data, etc.
- Ethical considerations
- Quality assurance and safety
- Formulation of hypothesis
- Objectives of the trial
- Sample size calculations
- Definition of the reference population
- Choice of a comparison treatment - placebo or current available best treatment
- Selection of intervention and control groups including: source, inclusion and exclusion criteria and methods of recruitment
- Informed consent procedures
- Choice of baseline measurements - include all variables considered or known to affect the outcome(s) of interest
- Method of random allocation of study participants to defined treatment groups (intervention vs. control)
- Follow-up of all treatment groups and assessment of outcomes
- Monitoring compliance and losses to follow-up
- Interim and final analysis plans
- Publication plans
The gold standard of intervention studies is the randomised, double-blind placebo-controlled trial. This attempts to reduce bias in the following ways:3
- Selection bias – reduced by random allocation to treatment-arms, so each participant has an equal chance of receiving the intervention.
- Blinding – reduces measurement bias because both the investigators and the participants (hence ‘double-blind’) are unaware of the treatment allocations, so this cannot affect how they are assessed.
- Randomisation also ensures that the control group is as similar to the intervention group as possible, i.e. that confounders are equally distributed between the groups.
Randomisation
The aim of randomisation is to ensure that any observed differences between the study groups are due to differences in the treatment alone and not due to the effects of confounding or bias. Randomisation ensures that trial groups are similar at the outset in all respects with the exception of the intervention under investigation (i.e. they are balanced). Note that there still may be differences in how the groups are handled as the trial progresses (introducing performance bias).
Methods of random allocation are used to ensure that all study participants have the same chance of allocation to the treatment or control group, and that the likelihood of receiving an intervention is equal regardless of when the participant entered the study. Therefore, the probability of any participant receiving the intervention should be independent of any other participant being assigned that treatment.
The assignment of study subjects to each intervention is determined by formal chance processes and cannot be predicted or influenced by the investigator or participant. In a well-designed RCT, the random allocation sequence is pre-determined and cannot be influenced. It is important that those responsible for recruiting participants into a study are unaware which study arm the individual will be allocated to. Allocation concealment avoids both conscious and unconscious selection of patients into the study, and attempts to ensure that the investigators cannot manipulate the trial by influencing which arm participants are enrolled into. One method of achieving this is by concealing allocation details in sealed, opaque envelopes. An alternative (and better) method is central randomisation by telephone, where the clinician calls a randomisation service to get the treatment allocation. The latter allocation mechanism is beyond the control of both the investigator and the participant, limiting potential bias in allocating the treatment. The process should be carried out once the participant has been determined to be eligible for inclusion and after they have given informed consent to participate.
Methods of treatment allocation
- Systematic allocation
Participants can be allocated to study groups alternately, or on alternate days (“alternation”). Another method would be allocation on the basis of date of birth (“quasi-alternation”). These methods are not considered to be truly random and can result in selection bias. It may be possible for the investigator to manipulate the process, if they favour one study arm over another.
- Simple randomisation
For example, using computer generated random number sequences (i.e. odd and even numbers can be used to allocate to one of two trial arms) or tossing a coin. Simple randomisation can suffer from chance bias – where the resulting groups are not balanced in important potential confounders by chance.
- Block randomisation
Block randomisation is a method used to ensure that the numbers of participants assigned to each group is equally distributed and is commonly used in smaller trials. Participants are frequently allocated in blocks of 4, for example, so it is guaranteed that 2 in each block will be randomised to each study group. Other block sizes can be used, and block sizes can be varied within a trial. With blocks of 4, there are only six possible sequences to allocate two to group A and two to group B (AABB, ABAB, BABA, etc …) and blocks can be chosen at random to generate the allocation sequence. Large block sizes should be avoided, as this increases the chances of ending up with unequal group sizes. For example, a block size of 16 could have the sequence AAABAABAAABBBBBB. If the study was terminated part-way through this block, there would be a clear imbalance in the group sizes in favour of group A. Investigators should not know the block size, to prevent them from predicting allocation for the last few participants in each block.
- Stratified randomisation
Stratified randomisation is used to ensure that potential confounding factors thought to be associated with the outcome are evenly distributed between groups.2 Prior to randomisation, participants are separated into different subgroups or strata based on key risk factors that may influence outcome, for example sex or age. Individuals are then randomly allocated to each treatment arm from within the strata. This method of treatment allocation should be based on block randomisation within each stratum, to ensure there are similar numbers of participants in each arm. However, there are a limited number of baseline variables that can be balanced by stratification because of the potential for small numbers of subjects within each stratum.
- Minimisation
Although technically not a random method, minimisation may be used in small trials where other methods of randomisation will not result in balanced groups. The treatment allocated to the next participant being enrolled in the trial depends on the characteristics and allocation of the participants already enrolled. The aim is that important potential confounding factors are distributed across the groups as equally as possible.
Once allocation is complete, the success of the randomisation process should be confirmed by comparing baseline factors between the two groups, to ensure that they are similar.
Advantages of randomisation
- Eliminates confounding - tends to create groups that are comparable for all factors that may influence outcome (known, unknown or difficult to measure). Therefore, the only difference between the groups should be the intervention.
- Eliminates treatment selection bias.
- Gives validity in statistical tests based on probability theory.
- Any baseline differences that exist between study groups are attributable to chance rather than bias - though this should still be considered as a potential concern.
Disadvantages of randomisation
- Does not guarantee comparable groups as differences in confounding variables may arise by chance.
Blinding in randomised controlled trials
Blinding is used in RCTs to ensure that there are no differences in the way the study arms are assessed or managed, thus minimising bias. Bias may be introduced, for example, if the investigator is aware which treatment a subject is receiving, as this may influence (intentionally or unintentionally) the way in which they measure or interpret the outcome data. Similarly, a subject's knowledge of treatment assignment may influence their response to a specific treatment.
Blinding also involves ensuring that the intervention and standard/placebo treatment appears the same. However, blinding is not always possible, for example when the treatment involved physiotherapy exercises, a weight loss programme or counselling.
In a double-blind trial, neither the investigator nor the study participant are aware of treatment assignments. However, this design is not always feasible and a single-blind RCT is where the investigator, but not the study participant, knows which treatment has been allocated.
Variations of the randomised control trial
- Crossover trials
In a crossover trial each subject acts as their own control, and receives all the treatments under investigation in sequence. Random allocation determines the sequence in which each participant receives the treatments. This type of trial may be used when the intervention does not have long-term effects. A particular advantage of a crossover trial is that the groups are as similar as possible. In crossover trials it is common to have a 'washout period' where no treatment is given between each intervention to avoid the possibility of a residual effect from the previous treatment.
- Factorial trials
A factorial trial is where two or more interventions are evaluated singly and simultaneously compared with a control group in the same trial. For example, where two interventions are being tested (A and B) along with a control, this would result in four groups: control, A only, B only, and both A and B. This type of RCT is commonly used to evaluate interactions between interventions.
- Cluster-randomised controlled trials
Cluster randomised trials involve groups of individuals or communities, as opposed to individuals. Groups or communities are randomised to receive the intervention or standard/no treatment. This type of study design may be used to evaluate preventative health services such as smoking cessation programmes. For example, all patients attending a single general practice may be allocated to receive the new programme, whilst those at another practice will receive standard care. In this sort of trial, intra-cluster correlation needs to be taken into account in assessing effect size and sample size (see chapter “Clustered data”).
Ethical issues
The use of RCTs raises important ethical issues. For example, there must be sufficient doubt about the particular agent being tested to allow withholding of it from half the subjects, and at the same time there must be sufficient belief in the agent's potential to justify exposing the remaining half of all willing and eligible participants.1 This is known as clinical equipoise.
In addition, there must be sufficient belief that the intervention under investigation is safe.
Informed consent is essential in RCTs (as it is in other study designs). Study subjects must understand that they are participating in an experiment and that in a placebo-controlled trial they may receive an inactive product. In addition, participants must be informed of the aims, methods and potential benefits or hazards of participating in the trial.
In a controlled trial careful consideration should also be given to what intervention is given to the control group. For example, if an effective treatment already exists, participants in the control group should not receive a placebo, depriving them of treatment. Any comparison should therefore be between the current standard treatment and the new treatment.
It is essential that study participants do not suffer as a consequence of a RCT. Most RCTs incorporate a data monitoring committee who are independent of the investigators, whose function is to review safety and efficacy data, and to ensure quality and compliance.
Analysis of RCTs
The analysis of RCT data is focused on estimating the size of the difference in predefined outcomes between the intervention and control groups. The main measure of effect obtained is the rate or risk ratio.
For trials of preventative interventions, the protective efficacy (or effectiveness) is calculated as:

(where R is the risk or rate)
Intention-to-treat analysis
When RCTs are analysed using an intention-to-treat analysis (ITT), participants’ results are analysed in the group to which they were originally assigned.5 This should happen regardless of whether they were lost to follow-up, or whether they switched treatment groups during the trial. During the RCT subjects may refuse to continue to participate and stop taking their allocated treatment.
If the investigators exclude participants from the analysis where they have not adhered to their allocated treatment strategy, the estimate of the effect of the treatment is likely to be flawed. The aim of the intention-to-treat analysis is to provide a pragmatic (real-world) estimate of the benefit of the treatment under investigation, rather than of its potential benefit in patients who receive treatment exactly as planned. Subjects who failed to take their allocated treatment may have done so because of adverse side effects, or because they felt it was not working, and - in the real world - patients do not always take their medication as prescribed.
Additionally, there is evidence to suggest that those participants who are fully compliant with their prescribed medication do better than those who do not adhere, even after adjustment for all known prognostic factors and irrespective of assignment to active treatment or placebo.
Excluding non-compliant participants from the analysis leaves those who may be destined to have a better outcome, and destroys the unbiased comparison afforded by randomisation.6 Where a comparison is performed using only those participants who completed the treatment originally allocated, this is known as a per protocol analysis.
Where full outcome data is not known for all subjects, then researchers can either impute an outcome for each individual lost based on shared characteristics with other individuals in the study, or use the last recorded measure.
In summary, during the statistical analysis of an RCT, all study subjects should ideally be retained in the group to which they were originally allocated regardless of whether or not that was the treatment received.
Strengths of randomised controlled trials
A well designed RCT provides the strongest epidemiological evidence of any study design about the effectiveness and safety of a given intervention.
- An RCT is considered to be the best type of epidemiological study from which to draw conclusions about causality.
- Randomisation is a powerful tool for controlling for confounding, even for factors that may be unknown or difficult to measure. If a study is designed and conducted rigorously, the possibility that an observed association is due to confounding should be minimal.
- Provides a strong basis for statistical inference.
- Enables blinding and therefore minimises bias.
- Can measure disease incidence and multiple outcomes.
Weaknesses of randomised controlled trials
- Ethical constraints.
- Expensive and time consuming.
- Requires complex design and analysis if unit of allocation is not the individual.
- Inefficient for rare diseases or diseases with a delayed outcome.
- There may be a placebo effect in the non-intervention group, which may result in an underestimation of the true treatment effect.
- Short follow up periods; not good at assessing long-term outcomes or harms.
- Generalisability: subjects in an RCT may be more willing to comply with the treatment regimen and therefore may not be representative of all individuals in a population eligible for the treatment.
Examples of RCTs
One of the first randomised trials to be carried out was the evaluation of streptomycin in the treatment of tuberculosis, published in 1948. Since then, there have been numerous other examples which have had a significant impact on clinical practice.
A non-randomised study published in 1980 evaluated the possible benefit of vitamin supplementation at the time of conception in women at high risk of having a baby with a neural tube defect. The investigators found that the vitamin group subsequently had fewer babies with neural tube defects than the placebo control group. The control group included women ineligible for the trial as well as women who refused to participate. As a consequence, the findings were not widely accepted, and the Medical Research Council later funded a large RCT to answer the question in a way that would be widely accepted.7
- Streptomycin treatment of pulmonary tuberculosis: a Medical Research Council investigation. BMJ 1948;2:769-782.
- MRC Vitamin Study Research Group. Prevention of neural tube defects: results of the Medical Research Council vitamin study. Lancet 1991; 338: 131-137
References
- Hennekens CH, Buring JE. Epidemiology in Medicine, Lippincott Williams & Wilkins, 1987.
- http://www.edmundjessop.org.uk/fulltext.doc - Accessed 20/12/16
- Carneiro I, Howard N. Introduction to Epidemiology. Open University Press, 2011.
- Kendall JM. Designing a research project: Randomised Controlled trials and their principles,Emerg Med J. 2003, March;20(2)164-168.
- Hollis S, Campbell F, What is meant by intention to treat analysis? Survey of publishedrandomised controlled trials. BMJ 1999; 319;670-74.
- Montori V, Guyatt G. Intention-to-treat principle. CMAJ 2001; 165 (10)
- Altman D, Bland M. Treatment allocation in controlled trials: why randomise? BMJ 1999;318:1209-1209
Further Resources
- Pocock SJ. Clinical Trials: A practical approach, Chichester, Wiley, 1984.
- Sibbald B, Roland M, Understanding controlled trials: Why are randomised controlled trials important?, BMJ 1998, 316:201.
- Altman DG, Randomisation. BMJ 1991;302;1481-2.
© Helen Barratt, Maria Kirwan 2009, Saran Shantikumar 2018